packages <- c("igraph", "tidyverse", "devtools",
"R.utils","vegan","vctrs","magrittr")
install.packages(setdiff(packages,
rownames(installed.packages())),
dependencies = TRUE
)DNA metabarcoding diet analysis in reindeer is quantitative and integrates feeding over several weeks
Stefaniya Kamenova 
Eric Coissac
Abstract
Filtering of the SPER01 DNA metabarcoding raw data.
Setting up the R environment
Install missing packages
Loading of the R libraries
ROBIToolspackage is used to read result files produced by OBITools.ROBITaxonomypackage provides function allowing to query OBITools formated taxonomy.
if (!"ROBITools" %in% rownames(installed.packages())) {
# ROBITools are not available on CRAN and have to be installed
# from http://git.metabarcoding.org using devtools
metabarcoding_git <- "https://git.metabarcoding.org/obitools"
devtools::install_git(paste(metabarcoding_git,
"ROBIUtils.git",
sep="/"))
devtools::install_git(paste(metabarcoding_git,
"ROBITaxonomy.git",
sep="/"))
devtools::install_git(paste(metabarcoding_git,
"ROBITools.git",
sep="/"))
}
library(ROBITools)
library(ROBITaxonomy)tidyverse(Wickham et al., 2019) provides various method for efficient data manipulation and plotting viaggplot2(Wickham, 2016)
library(tidyverse)library(R.utils)library(vegan)library(magrittr)source("methods.R")
Attaching package: 'matrixStats'The following object is masked from 'package:dplyr':
count
Attaching package: 'vctrs'The following object is masked from 'package:dplyr':
data_frameThe following object is masked from 'package:tibble':
data_frameLoading the data
Load the NCBI taxonomy
if (! file.exists("Data/ncbi20210212.adx")) {
gunzip("Data/ncbi20210212.adx.gz",remove=FALSE)
gunzip("Data/ncbi20210212.ndx.gz",remove=FALSE)
gunzip("Data/ncbi20210212.rdx.gz",remove=FALSE)
gunzip("Data/ncbi20210212.tdx.gz",remove=FALSE)
}taxo <- read.taxonomy("Data/ncbi20210212")Loading the metabarcoding data
if (! file.exists("Data/Rawdata/SPER01_all_paired.ali.assigned.ann.diag.uniq.ann.c1.l10.clean.EMBL.tag.ann.sort.uniq.nosingleton.tab"))
gunzip("Data/Rawdata/SPER01_all_paired.ali.assigned.ann.diag.uniq.ann.c1.l10.clean.EMBL.tag.ann.sort.uniq.nosingleton.tab.gz",remove=FALSE)
SPER01.raw = import.metabarcoding.data("Data/Rawdata/SPER01_all_paired.ali.assigned.ann.diag.uniq.ann.c1.l10.clean.EMBL.tag.ann.sort.uniq.nosingleton.tab.gz")Loading the metadata
samples.metadata = read_csv("Data/Faeces/sampling_dates.csv",
show_col_types = FALSE)Sample description
Normalization of samples names
Extract information relative to PCR replicates and sample names.
sample_names_split = strsplit(as.character(sample_names), "_R")
replicate = sapply(sample_names_split, function(x) x[length(x)])
sample_id = sapply(sample_names_split, function(x) x[1])
samples_desc = data.frame(name = samples(SPER01.raw)$sample, replicate = replicate, sample_id = sample_id)
SPER01.raw@samples = samples_descCategorize MOTUs
DNA Sequence of the 6 synthetic sequences used as SPER01 positive controls.
Standard1 = "taagtctcgcactagttgtgacctaacgaatagagaattctataagacgtgttgtcccat"
Standard2 = "gtgtatggtatatttgaataatattaaatagaatttaatcaatctttacatcgcttaata"
Standard3 = "cacaatgctcggtaactagaagcatttgta"
Standard4 = "attgaatgaaaagattattcgatatagaat"
Standard5 = "agaacgctagaatctaagatggggggggggatgagtaagatatttatcagtaacatatga"
Standard6 = "atttttgtaactcattaacaattttttttttgatgtatcataagtactaaactagttact"- Identify which MOTUs are corresponding to these positive control sequences and associated them to their corresponding category.
- All the MOTUs exhibiting a similarity with one of the reference SPER01 database greater than 95% is tagged as
SPER01 - The remaining sequences are tagged as
Unknown
sequence_type = rep("Unknown", nrow(motus(SPER01.raw)))
sequence_type[which(motus(SPER01.raw)$`best_identity:db_GH`> 0.95)] = "SPER01"
sequence_type[which(motus(SPER01.raw)$sequence == Standard1)] = "standard1"
sequence_type[which(motus(SPER01.raw)$sequence == Standard2)] = "standard2"
sequence_type[which(motus(SPER01.raw)$sequence == Standard3)] = "standard3"
sequence_type[which(motus(SPER01.raw)$sequence == Standard4)] = "standard4"
sequence_type[which(motus(SPER01.raw)$sequence == Standard5)] = "standard5"
sequence_type[which(motus(SPER01.raw)$sequence == Standard6)] = "standard6"
SPER01.raw@motus$sequence_type = as.factor(sequence_type)
table(SPER01.raw@motus$sequence_type)
SPER01 standard1 standard2 standard3 standard4 standard5 standard6 Unknown
44125 1 1 1 1 1 1 36419 Curation procedure
Select motus occuring at least at 1% in at least one PCR
Only MOTUs occurring at least at one percent in at least one PCR are conserved. The others are discarded and correspond to few rare taxa, and many spurious MOTUs generated by PCR artefacts.
norare = apply(decostand(reads(SPER01.raw),method = "total"),
MARGIN = 2,
FUN = max) >= 0.01
table(norare)norare
FALSE TRUE
80356 194 SPER01.norare <- SPER01.raw[,which(norare)]SPER01.norare.reads_per_motu = colSums(reads(SPER01.norare))
plot(SPER01.norare.reads_per_motu,log="y",
cex=0.1 + 0.5 * (SPER01.norare@motus$sequence_type!="SPER01"),
col = as.integer(SPER01.norare@motus$sequence_type), pch=16)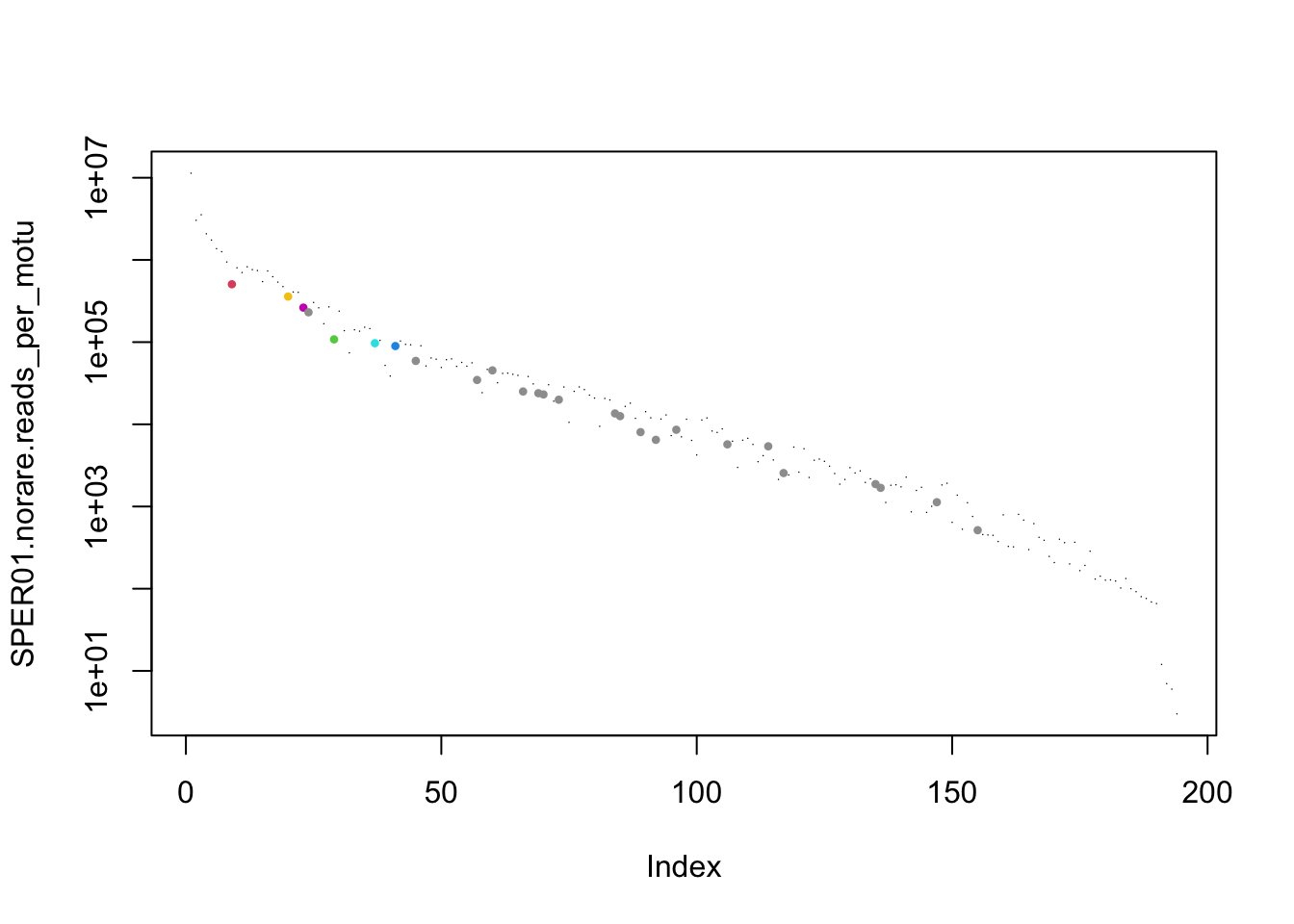
Analysis of the PCR Positive controls
Synthetic MOTUs are extracted from the data set and their relative read abundances (RRA) is plotted as a function of their theoretical abundances to check the quality of the PCR.
rp <- SPER01.norare.reads_per_motu[! SPER01.norare@motus$sequence_type %in% c("SPER01","Unknown")]
rp/sum(rp)GHP2_00000016 GHP3_00000014 GHP3_00000156 GHP2_00000010 GHP3_00000144
0.35574603 0.25211192 0.18491040 0.07594321 0.06826363
GHP2_00000144
0.06302481 expectedRRA <- 1/2^(1:6)
observedRRA <- rp/sum(rp)
plot(expectedRRA,observedRRA,
xlab="expected RRA",
ylab="observed RRA")
abline(lm(observedRRA ~ expectedRRA),
col = "blue", lty = 2)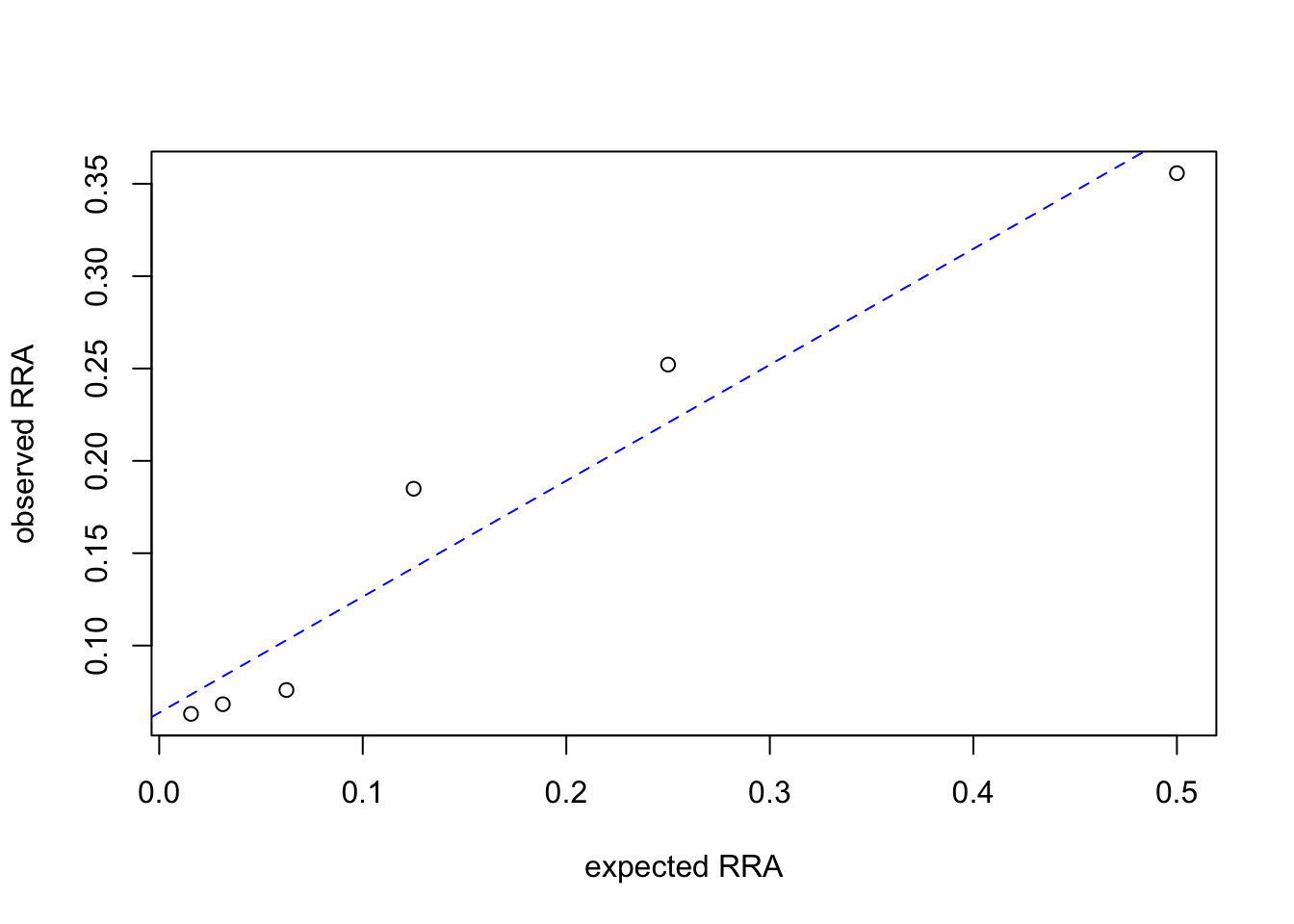
Filtering for PCR outliers
Only library 1 and 2 have individually tagged PCR replicates.
library_3.ids = read.csv("Data/samples_library_3.txt",
stringsAsFactors = FALSE,
header = FALSE)[,1]library3.keep = gsub("_R.?$","_R",rownames(SPER01.norare)) %in% library_3.ids
SPER01.lib3 = SPER01.norare[library3.keep,]
SPER01.lib12= SPER01.norare[!library3.keep,]
dim(SPER01.lib3)[1] 65 194dim(SPER01.lib12)[1] 541 194Load the script containing the selection procedure implemented in function tag_bad_pcr.
source("select_pcr.R")First selection round
keep1 = tag_bad_pcr(samples = samples(SPER01.lib12)$sample_id,
counts = reads(SPER01.lib12),
plot = TRUE
)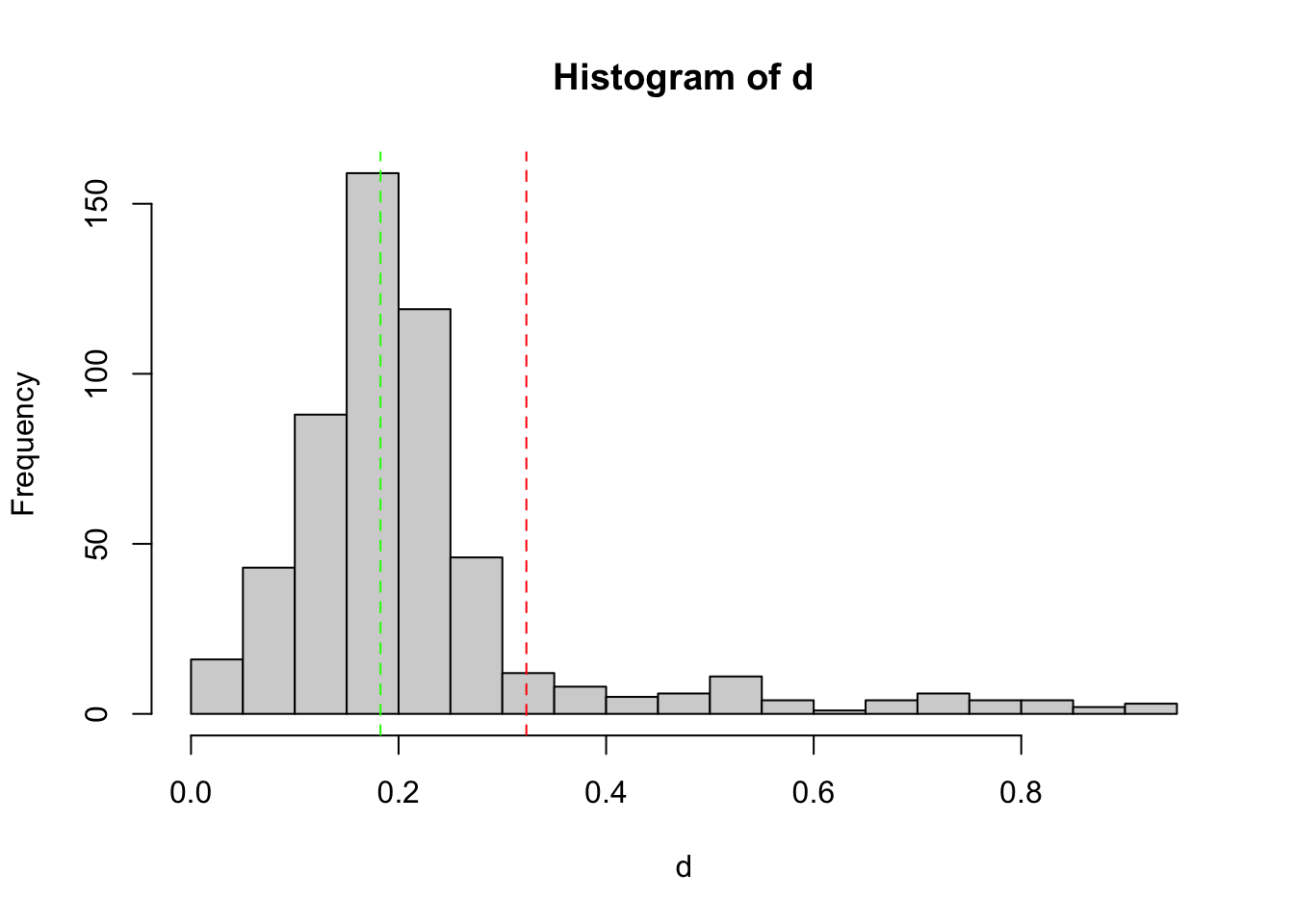
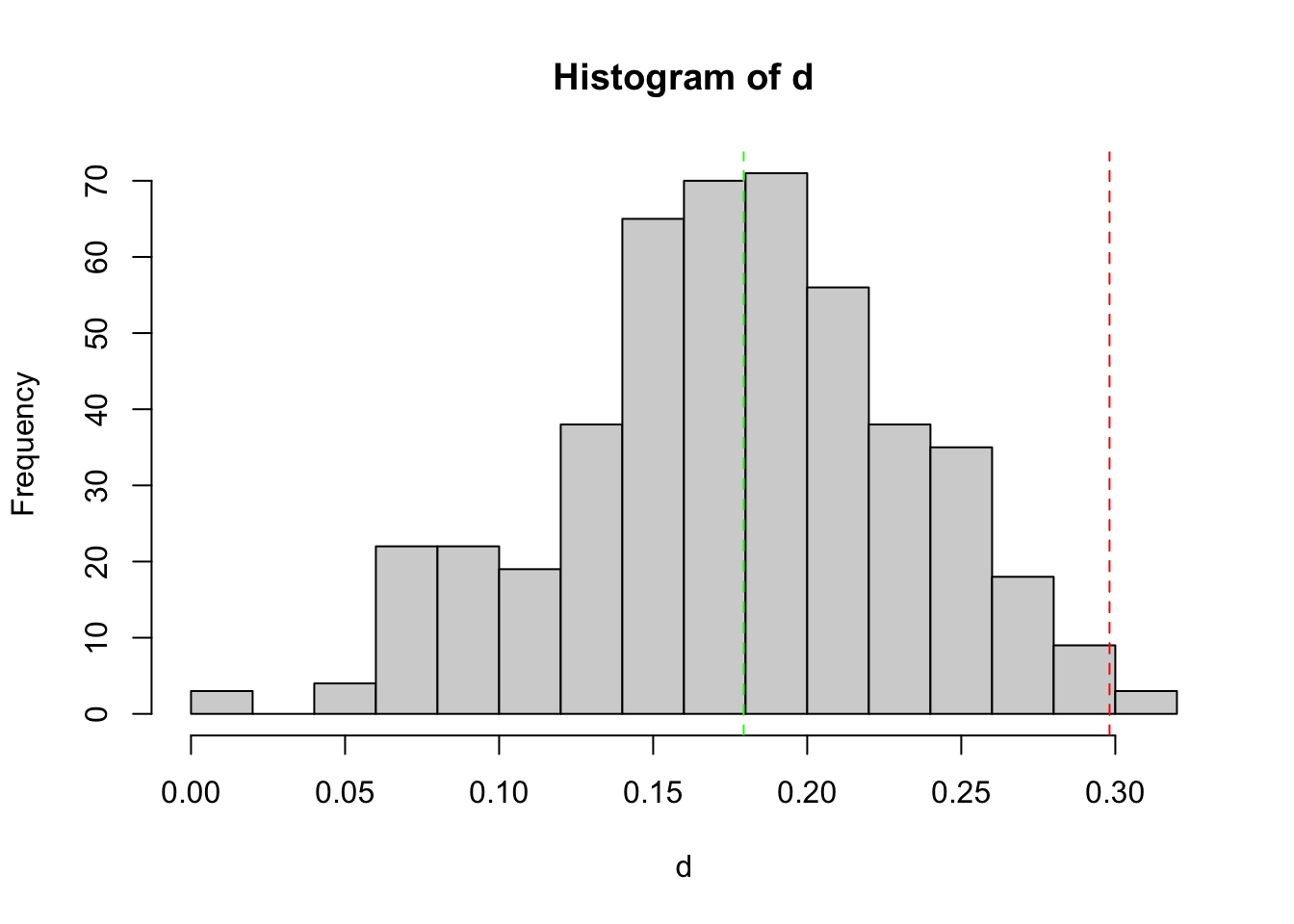
Histogram shows the empirical distribution of the PCR replicate distances. The red vertical dashed line indicates the threshold used to discard outlier PCRs. The green vertical dashed line indicates the mode of the observed distribution.
table(keep1$keep)
FALSE TRUE
44 497 FALSEis the count of PCR to discard, TRUE the count of PCR conserved at the end of this selection round.
samples(SPER01.lib12)$name[!keep1$keep] [1] "DNANC12_R3" "DNANC14_R3" "DNANC15_R3" "DNANC_10_R2" "DNANC_11_R3"
[6] "DNANC_13_R3" "DNANC_14_R3" "DNANC_15_R3" "DNANC_7_R2" "DNANC_8_R2"
[11] "DNANC_9_R2" "PCRNC_3_R3" "PCRNC_6_R3" "PCRPOS_3_R3" "PCRPOS_4_R2"
[16] "PCRPOS_5_R3" "PCRPOS_6_R3" "X_29_R1" "X_38_R3" "X_50_R2"
[21] "X_64_R1" "X_66_R1" "Y_24_R2" "Y_28_R2" "Y_2_R1"
[26] "Y_33_R2" "Y_36_R2" "Y_44_R1" "Y_45_R1" "Y_46_R3"
[31] "Y_47_R1" "Y_48_R1" "Y_48_R2" "Y_49_R2" "Y_51_R2"
[36] "Y_52_R1" "Y_56_R3" "Z_20_R1" "Z_30_R1" "Z_36_R1"
[41] "Z_45_R2" "Z_53_R1" "Z_5_R1" "Z_61_R3" Above is the list of the ids of the discarded PCRs.
SPER01.lib12.k1 = SPER01.lib12[keep1$keep,]Second selection round
keep2 = tag_bad_pcr(samples = samples(SPER01.lib12.k1)$sample_id,
counts = reads(SPER01.lib12.k1),
plot = TRUE
)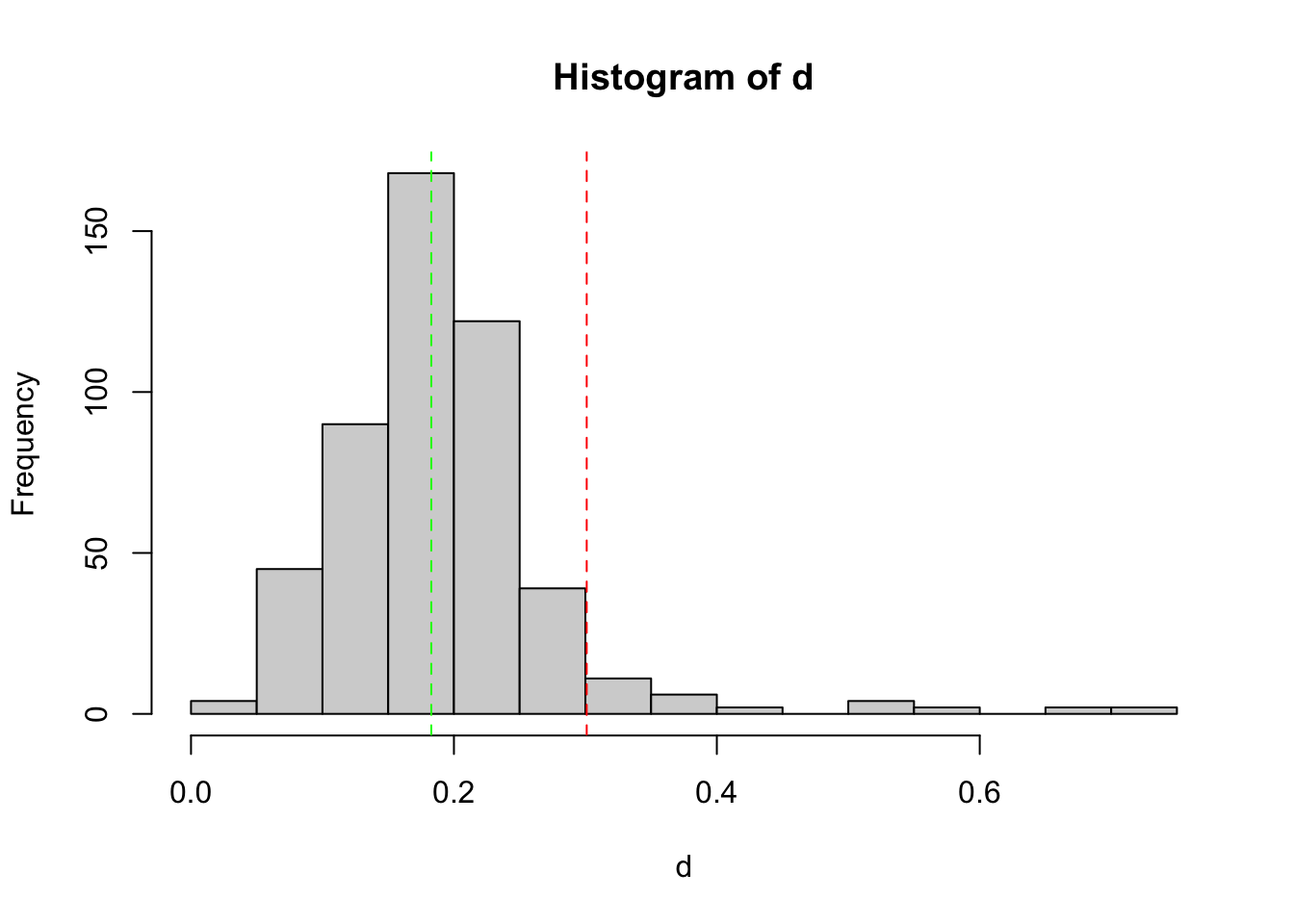
table(keep2$keep)
FALSE TRUE
24 473 samples(SPER01.lib12.k1)$name[!keep2$keep] [1] "PCRPOS_3_R2" "X_50_R1" "X_50_R3" "Y_18_R1" "Y_2_R2"
[6] "Y_2_R3" "Y_31_R1" "Y_44_R2" "Y_46_R1" "Y_46_R2"
[11] "Y_47_R2" "Y_49_R1" "Y_49_R3" "Y_56_R1" "Y_56_R2"
[16] "Y_71_R1" "Z_30_R2" "Z_30_R3" "Z_45_R1" "Z_49_R1"
[21] "Z_53_R2" "Z_53_R3" "Z_61_R1" "Z_61_R2" SPER01.lib12.k2 = SPER01.lib12.k1[keep2$keep,]Third selection round
keep3 = tag_bad_pcr(samples = samples(SPER01.lib12.k2)$sample_id,
counts = reads(SPER01.lib12.k2),
plot = TRUE
)
table(keep3$keep)
FALSE TRUE
7 466 keep3[!keep3$keep,] samples distance maximum repeats keep
X_14_R3 X_14 0.2982691 0.2982691 3 FALSE
Y_44_R3 Y_44 0.0000000 0.0000000 1 FALSE
Y_47_R3 Y_47 0.0000000 0.0000000 1 FALSE
Y_50_R3 Y_50 0.2986271 0.2986271 3 FALSE
Y_52_R3 Y_52 0.3004914 0.3004914 2 FALSE
Z_45_R3 Z_45 0.0000000 0.0000000 1 FALSE
Z_55_R3 Z_55 0.3004595 0.3004595 3 FALSESPER01.lib12.k3 = SPER01.lib12.k2[keep3$keep,]Merge remaining PCR replicates
freq = decostand(reads(SPER01.lib12.k3),
method = "total")
SPER01.lib12.k3$count = reads(SPER01.lib12.k3)
SPER01.lib12.k3@reads = freq
SPER01.merged = aggregate(SPER01.lib12.k3, MARGIN = 1, by = list(sample_id=samples(SPER01.lib12.k3)$sample_id), FUN = mean)Look for controls left in library 1 and 2
rownames(SPER01.merged) [1] "X_10" "X_11" "X_12" "X_14" "X_15" "X_16" "X_17" "X_18" "X_19" "X_2"
[11] "X_20" "X_21" "X_22" "X_23" "X_24" "X_25" "X_26" "X_27" "X_28" "X_29"
[21] "X_3" "X_30" "X_31" "X_33" "X_34" "X_35" "X_36" "X_37" "X_38" "X_39"
[31] "X_4" "X_41" "X_42" "X_44" "X_51" "X_53" "X_54" "X_56" "X_57" "X_59"
[41] "X_60" "X_63" "X_64" "X_65" "X_66" "X_68" "X_70" "X_74" "X_75" "X_76"
[51] "X_77" "X_78" "X_79" "X_80" "X_9" "Y_1" "Y_11" "Y_13" "Y_14" "Y_18"
[61] "Y_21" "Y_23" "Y_24" "Y_25" "Y_26" "Y_28" "Y_29" "Y_3" "Y_31" "Y_32"
[71] "Y_33" "Y_34" "Y_36" "Y_38" "Y_39" "Y_4" "Y_40" "Y_41" "Y_42" "Y_43"
[81] "Y_45" "Y_5" "Y_50" "Y_51" "Y_52" "Y_53" "Y_57" "Y_58" "Y_59" "Y_6"
[91] "Y_61" "Y_69" "Y_7" "Y_70" "Y_71" "Y_72" "Y_74" "Y_8" "Y_9" "Z_1"
[101] "Z_10" "Z_11" "Z_12" "Z_13" "Z_14" "Z_15" "Z_16" "Z_17" "Z_18" "Z_19"
[111] "Z_21" "Z_22" "Z_23" "Z_24" "Z_25" "Z_27" "Z_28" "Z_3" "Z_31" "Z_32"
[121] "Z_33" "Z_34" "Z_35" "Z_36" "Z_37" "Z_38" "Z_4" "Z_40" "Z_42" "Z_43"
[131] "Z_44" "Z_46" "Z_48" "Z_49" "Z_5" "Z_51" "Z_52" "Z_54" "Z_55" "Z_56"
[141] "Z_59" "Z_6" "Z_60" "Z_62" "Z_63" "Z_65" "Z_66" "Z_67" "Z_68" "Z_69"
[151] "Z_7" "Z_70" "Z_71" "Z_72" "Z_73" "Z_74" "Z_75" "Z_76" "Z_77" "Z_78"
[161] "Z_79" "Z_8" "Z_80"Merge lib 1,2 and 3
Remove controls in library 3
rownames(SPER01.lib3) [1] "DNANC_3_R1" "DNANC_4_R1" "DNANC_5_R1" "DNANC_6_R1" "DNANC_6_R2"
[6] "PCRNC_1_R1" "PCRNC_2_R1" "PCRNC_2_R2" "PCRPOS_1_R1" "PCRPOS_2_R1"
[11] "PCRPOS_2_R2" "X_1_R1" "X_32_R1" "X_40_R1" "X_43_R1"
[16] "X_45_R1" "X_46_R1" "X_47_R1" "X_48_R1" "X_49_R1"
[21] "X_52_R1" "X_55_R1" "X_58_R1" "X_5_R1" "X_62_R1"
[26] "X_67_R1" "X_69_R1" "X_6_R1" "X_71_R1" "X_72_R1"
[31] "X_73_R1" "X_7_R1" "X_8_R1" "Y_10_R1" "Y_12_R1"
[36] "Y_15_R1" "Y_16_R1" "Y_17_R1" "Y_19_R1" "Y_20_R1"
[41] "Y_22_R1" "Y_27_R1" "Y_30_R1" "Y_35_R1" "Y_37_R1"
[46] "Y_54_R1" "Y_55_R1" "Y_60_R1" "Y_62_R1" "Y_63_R1"
[51] "Y_64_R1" "Y_65_R1" "Y_66_R1" "Y_67_R1" "Y_68_R1"
[56] "Y_73_R1" "Z_26_R1" "Z_29_R1" "Z_2_R1" "Z_41_R1"
[61] "Z_47_R1" "Z_50_R1" "Z_57_R1" "Z_58_R1" "Z_9_R1" SPER01.lib3.samples = SPER01.lib3[-(1:11),]
rownames(SPER01.lib3.samples@reads) = sub("_R.?$","",rownames(SPER01.lib3.samples))
rownames(SPER01.lib3.samples) [1] "X_1" "X_32" "X_40" "X_43" "X_45" "X_46" "X_47" "X_48" "X_49" "X_52"
[11] "X_55" "X_58" "X_5" "X_62" "X_67" "X_69" "X_6" "X_71" "X_72" "X_73"
[21] "X_7" "X_8" "Y_10" "Y_12" "Y_15" "Y_16" "Y_17" "Y_19" "Y_20" "Y_22"
[31] "Y_27" "Y_30" "Y_35" "Y_37" "Y_54" "Y_55" "Y_60" "Y_62" "Y_63" "Y_64"
[41] "Y_65" "Y_66" "Y_67" "Y_68" "Y_73" "Z_26" "Z_29" "Z_2" "Z_41" "Z_47"
[51] "Z_50" "Z_57" "Z_58" "Z_9" Merge library 1, 2 and 3
SPER01.lib123.reads = rbind(SPER01.merged@reads,
decostand(SPER01.lib3.samples@reads,method = "total"))
common = intersect(names(SPER01.merged@samples),
names(SPER01.lib3.samples@samples))
SPER01.lib123.samples = rbind(SPER01.merged@samples[,common],
SPER01.lib3.samples@samples[,common])
SPER01.lib123 = metabarcoding.data(reads = decostand(SPER01.lib123.reads,method = "total"),
samples = SPER01.lib123.samples,
motus = SPER01.merged@motus)
dim(SPER01.lib123)[1] 217 194SPER01.lib123@samples$animal_id = sapply(SPER01.lib123@samples$sample_id,
function(x) strsplit(as.character(x),"_")[[1]][1])Check for empty MOTUs
Look at MOTUs still present in the data matrix, but represented by no more reads because of the filtering procedure.
zero = colSums(reads(SPER01.lib123)) == 0
table(zero)zero
FALSE TRUE
184 10 SPER01.nozero = SPER01.lib123[,!zero]table(SPER01.nozero@motus$sequence_type)
SPER01 standard1 standard2 standard3 standard4 standard5 standard6 Unknown
164 1 0 1 1 0 1 16 Filter out rare species
plot(SPER01.nozero@motus$`best_identity:db_GH`,
apply(reads(SPER01.nozero),2,max),
col=as.factor(SPER01.nozero@motus$sequence_type),
log="y",
ylab="max read fequency in a sample",
xlab="best identity with the reference database")
abline(h=0.01,col="red",lty=2)
abline(v=0.95,col="red",lty=2)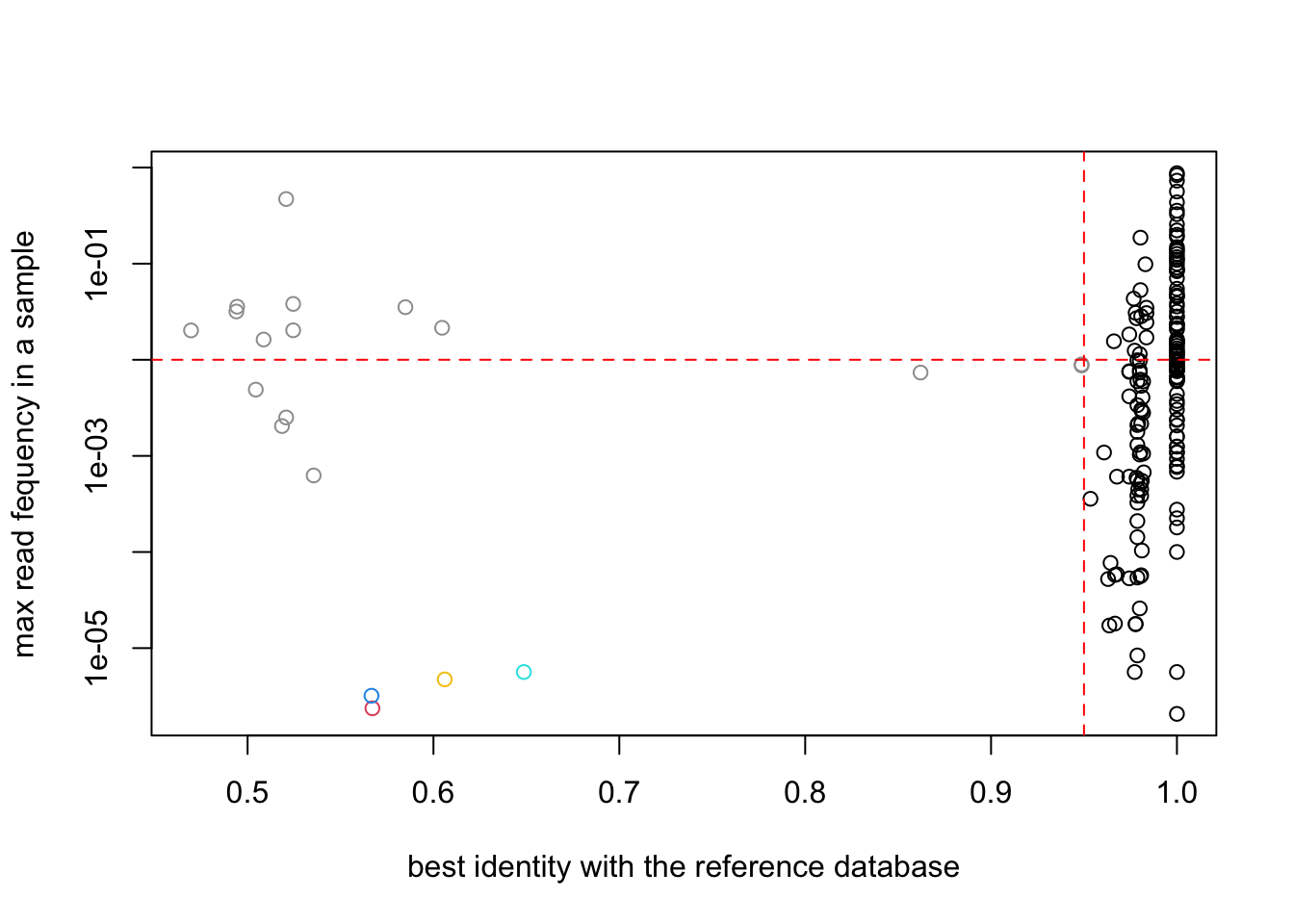
SPER01.merged3 = SPER01.lib123[, apply(reads(SPER01.lib123),2,max) > 0.01]plot(SPER01.merged3$motus$`best_identity:db_GH`,
apply(reads(SPER01.merged3),2,max),
col=as.factor(SPER01.merged3$motus$sequence_type),
log="y",
ylab="max read fequency in a sample",
xlab="best identity with the reference database")
abline(h=0.01,col="red",lty=2)
abline(v=0.95,col="red",lty=2)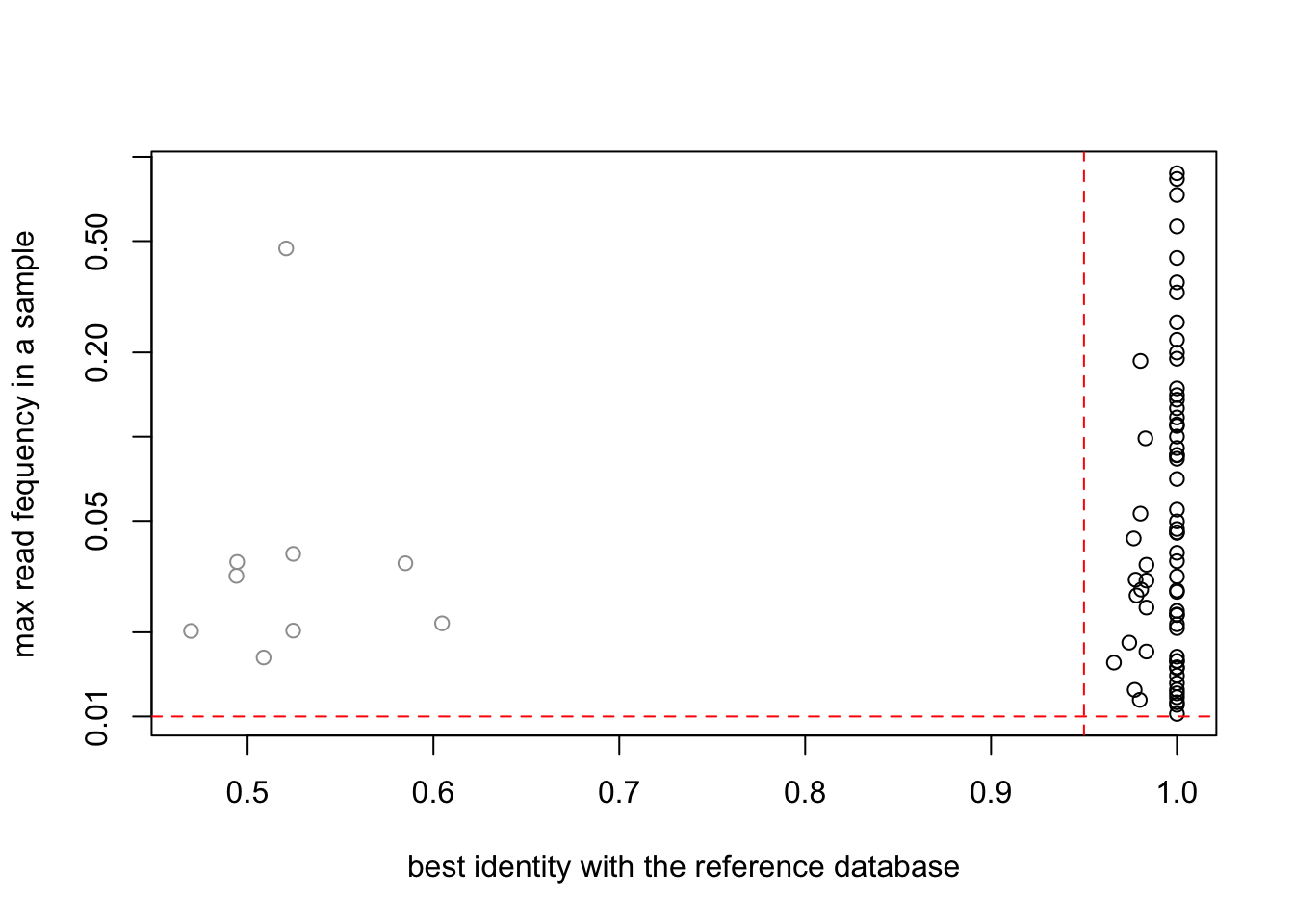
which(SPER01.merged3@motus$sequence_type == "Unknown" & apply(reads(SPER01.merged3),2,max) > 0.4)GHP3_00000038
21 SPER01.merged3@motus[21,"sequence"][1] "tatagggttttcttggtgtatttcacaccgaaccaggatgggcatgcaaaacaggttggtctcggtagttcagccctcgccatcggcaggggattttctacggctacatacatcgcctgggcattcaggcatttcatgaaaaactctcgtgcaagtagcattttctcatatcgctcttgattgatctctcctttctgtaccatccacaccgatgatactttgcata"Looks like nothing at embl by blast
Keep only MOTUs Strictly identical to one of the reference sequence
First level stringency filter (95% identity)
SPER01.merged4 = SPER01.merged3[,SPER01.merged3@motus$`best_identity:db_GH` > 0.95]
SPER01.merged4@reads = decostand(SPER01.merged4@reads,method = "total")
SPER01.merged4@motus <- SPER01.merged4@motus %>% select(-starts_with("obiclean_status:"))plot(SPER01.merged4$motus$`best_identity:db_GH`,
apply(reads(SPER01.merged4),2,max),
col=as.factor(SPER01.merged4$motus$sequence_type),
log="y",
ylab="max read fequency in a sample",
xlab="best identity with the reference database")
abline(h=0.01,col="red",lty=2)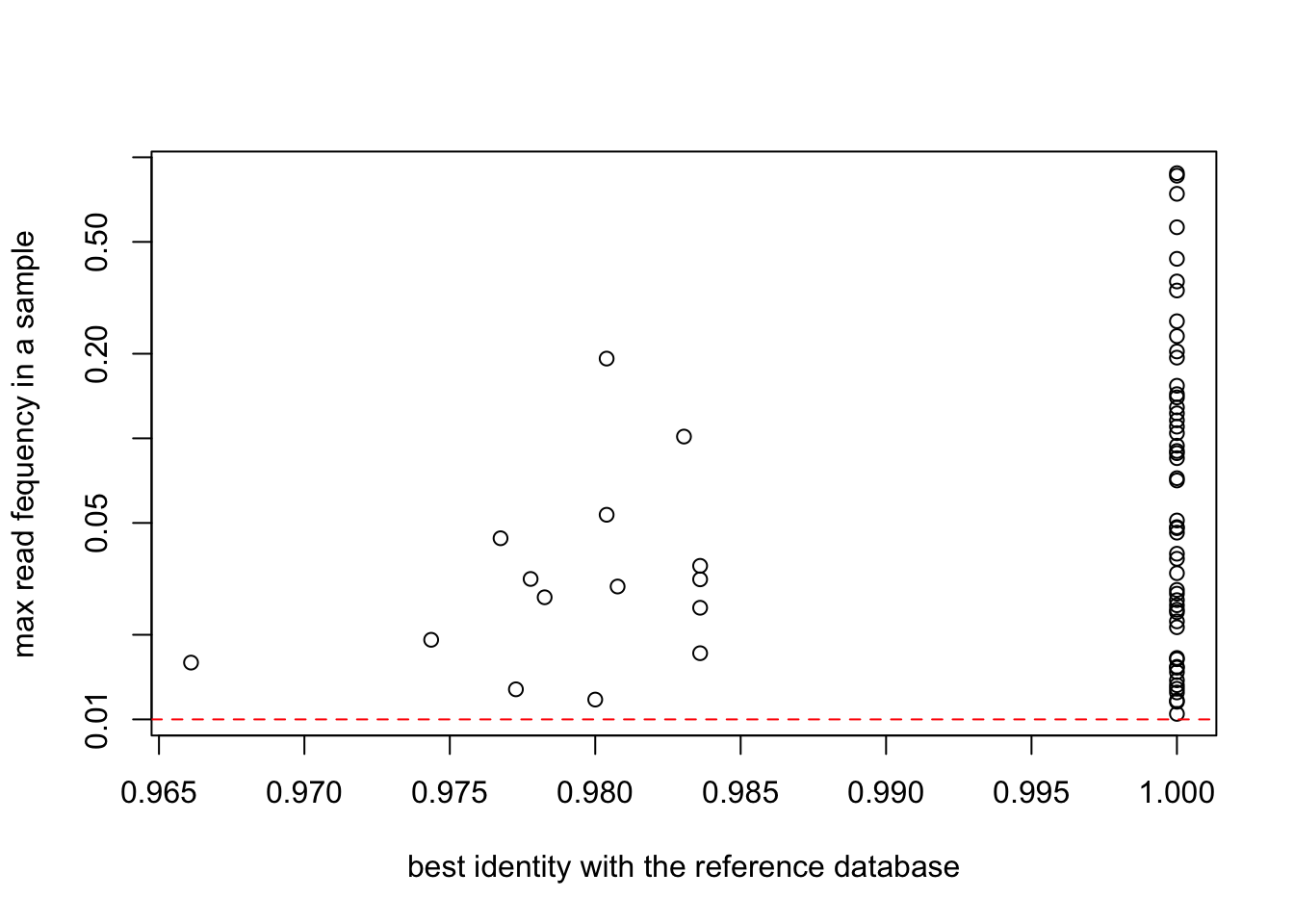
High stringency filtering (100% identity)
spermatophyta.taxid <- ecofind(taxo,patterns = "^Spermatophyta$")
SPER01.merged4@motus$is_spermatophyta <- is.subcladeof(taxo,SPER01.merged4@motus$taxid,spermatophyta.taxid)
table(SPER01.merged4@motus$is_spermatophyta)
FALSE TRUE
6 60 SPER01.merged4@motus %>% filter(!is_spermatophyta) id best_identity:db_GH best_match:db_GH count family
GHP1_00000082 GHP1_00000082 1 KY427333 137903 1203520
GHP1_00000361 GHP1_00000361 1 AF515231 49169 1203520
GHP3_00000569 GHP3_00000569 1 AF192562 56685 13803
GHP3_00000419 GHP3_00000419 1 HE993635 22646 1203500
GHP1_00000874 GHP1_00000874 1 AJ133265 13008 3250
GHP1_00008815 GHP1_00008815 1 AF023727 2987 52989
family_name genus genus_name match_count:db_GH rank
GHP1_00000082 Athyriaceae 32109 Athyrium 1 species
GHP1_00000361 Athyriaceae 32109 Athyrium 1 genus
GHP3_00000569 Sphagnaceae 13804 Sphagnum 1 genus
GHP3_00000419 Cystopteridaceae 32115 Gymnocarpium 1 species
GHP1_00000874 Lycopodiaceae NA <NA> 1 subfamily
GHP1_00008815 Orthotrichaceae NA <NA> 1 family
scientific_name species species_list:db_GH
GHP1_00000082 Athyrium sinense 672195 ['Athyrium sinense']
GHP1_00000361 Athyrium NA []
GHP3_00000569 Sphagnum NA []
GHP3_00000419 Gymnocarpium dryopteris 32116 ['Gymnocarpium dryopteris']
GHP1_00000874 Lycopodioideae NA []
GHP1_00008815 Orthotrichaceae NA []
species_name taxid
GHP1_00000082 Athyrium sinense 672195
GHP1_00000361 <NA> 32109
GHP3_00000569 <NA> 13804
GHP3_00000419 Gymnocarpium dryopteris 32116
GHP1_00000874 <NA> 1965347
GHP1_00008815 <NA> 52989
sequence sequence_type
GHP1_00000082 atcttgtattattcggatgaatttcgggcgatgaggcga SPER01
GHP1_00000361 atcttgtattattcagatgaatttcgggcgatgaggcga SPER01
GHP3_00000569 atcttgttttcataacataaatgg SPER01
GHP3_00000419 atcttgtattactcaaatgaatttcgggcaatgaggcaa SPER01
GHP1_00000874 atcctgtttagcaaatggcgg SPER01
GHP1_00008815 atattattttatttaaaaataa SPER01
is_spermatophyta
GHP1_00000082 FALSE
GHP1_00000361 FALSE
GHP3_00000569 FALSE
GHP3_00000419 FALSE
GHP1_00000874 FALSE
GHP1_00008815 FALSEmusaceae.taxid <- ecofind(taxo,patterns = "^Musaceae$")
to_keep <- ! (is.subcladeof(taxo,SPER01.merged4@motus$taxid,musaceae.taxid) | SPER01.merged4@motus$taxid==musaceae.taxid ) &
is.subcladeof(taxo,SPER01.merged4@motus$taxid,spermatophyta.taxid) &
SPER01.merged4@motus$`best_identity:db_GH` == 1
table(to_keep)to_keep
FALSE TRUE
22 45 SPER01.merged4@motus %>% filter(!to_keep) id best_identity:db_GH best_match:db_GH count
GHP2_00000044 GHP2_00000044 0.9803922 AB817362 2080755
GHP2_00000355 GHP2_00000355 0.9807692 AF098856 303606
GHP2_00000136 GHP2_00000136 0.9767442 DQ359689 261620
GHP1_00000082 GHP1_00000082 1.0000000 KY427333 137903
GHP2_00000549 GHP2_00000549 0.9830508 KX872610 135866
GHP1_00000014 GHP1_00000014 0.9803922 AB817372 52144
GHP2_00000171 GHP2_00000171 0.9743590 AC183493 90296
GHP2_00000260 GHP2_00000260 0.9777778 EF440558 51279
GHP2_00000820 GHP2_00000820 0.9772727 AJ505541 61937
GHP1_00000361 GHP1_00000361 1.0000000 AF515231 49169
GHP2_00000554 GHP2_00000554 0.9782609 AJ430966 62557
GHP3_00000569 GHP3_00000569 1.0000000 AF192562 56685
GHP1_00000681 GHP1_00000681 0.9836066 AB979732 24268
GHP3_00000153 GHP3_00000153 0.9836066 AB979732 19101
GHP1_00003018 GHP1_00003018 0.9836066 AB979732 10572
GHP1_00000030 GHP1_00000030 0.9836066 AB979732 25179
GHP3_00000419 GHP3_00000419 1.0000000 HE993635 22646
GHP1_00000874 GHP1_00000874 1.0000000 AJ133265 13008
GHP2_00008966 GHP2_00008966 0.9661017 KX872610 7043
GHP2_00009529 GHP2_00009529 0.9800000 AY344156 6375
GHP1_00008815 GHP1_00008815 1.0000000 AF023727 2987
GHP3_00027523 GHP3_00027523 1.0000000 AB817687 2256
family family_name genus genus_name match_count:db_GH
GHP2_00000044 NA <NA> NA <NA> 33
GHP2_00000355 4210 Asteraceae NA <NA> 6
GHP2_00000136 3440 Ranunculaceae 3445 Ranunculus 1
GHP1_00000082 1203520 Athyriaceae 32109 Athyrium 1
GHP2_00000549 4479 Poaceae NA <NA> NA
GHP1_00000014 3745 Rosaceae NA <NA> 14
GHP2_00000171 3700 Brassicaceae NA <NA> 1
GHP2_00000260 3318 Pinaceae 3337 Pinus 2
GHP2_00000820 4136 Lamiaceae NA <NA> 2
GHP1_00000361 1203520 Athyriaceae 32109 Athyrium 1
GHP2_00000554 NA <NA> NA <NA> 15
GHP3_00000569 13803 Sphagnaceae 13804 Sphagnum 1
GHP1_00000681 3514 Betulaceae NA <NA> 1
GHP3_00000153 3514 Betulaceae NA <NA> 1
GHP1_00003018 3514 Betulaceae NA <NA> 1
GHP1_00000030 3514 Betulaceae NA <NA> 1
GHP3_00000419 1203500 Cystopteridaceae 32115 Gymnocarpium 1
GHP1_00000874 3250 Lycopodiaceae NA <NA> 1
GHP2_00008966 4479 Poaceae NA <NA> 11
GHP2_00009529 14101 Juncaceae 13578 Juncus 2
GHP1_00008815 52989 Orthotrichaceae NA <NA> 1
GHP3_00027523 4637 Musaceae NA <NA> 1
rank scientific_name species
GHP2_00000044 order Asterales NA
GHP2_00000355 subfamily Asteroideae NA
GHP2_00000136 genus Ranunculus NA
GHP1_00000082 species Athyrium sinense 672195
GHP2_00000549 <NA> <NA> NA
GHP1_00000014 tribe Maleae NA
GHP2_00000171 tribe Brassiceae NA
GHP2_00000260 subgenus Pinus NA
GHP2_00000820 tribe Mentheae NA
GHP1_00000361 genus Athyrium NA
GHP2_00000554 order Asterales NA
GHP3_00000569 genus Sphagnum NA
GHP1_00000681 family Betulaceae NA
GHP3_00000153 family Betulaceae NA
GHP1_00003018 family Betulaceae NA
GHP1_00000030 family Betulaceae NA
GHP3_00000419 species Gymnocarpium dryopteris 32116
GHP1_00000874 subfamily Lycopodioideae NA
GHP2_00008966 no rank Poeae Chloroplast Group 1 (Aveneae type) NA
GHP2_00009529 genus Juncus NA
GHP1_00008815 family Orthotrichaceae NA
GHP3_00027523 family Musaceae NA
species_list:db_GH
GHP2_00000044 []
GHP2_00000355 ['Lasthenia californica', 'Achillea pseudoaleppica']
GHP2_00000136 []
GHP1_00000082 ['Athyrium sinense']
GHP2_00000549 ['Agrostis tolucensis']
GHP1_00000014 ['Photinia loriformis', 'Docynia delavayi', 'Crataegus monogyna', 'Photinia benthamiana', 'Dichotomanthes tristaniicarpa', 'Chaenomeles sinensis', 'Malus doumeri']
GHP2_00000171 []
GHP2_00000260 ['Pinus resinosa']
GHP2_00000820 []
GHP1_00000361 []
GHP2_00000554 ['Cymbonotus lawsonianus', 'Saussurea nematolepis', 'Helichrysum glumaceum', 'Villarsia sp. Fay s.n.', 'Argophyllum sp. Telford 5462', 'Dimorphotheca sinuata', 'Anisopappus corymbosus', 'Chaetanthera sp. Moreira 1736']
GHP3_00000569 []
GHP1_00000681 []
GHP3_00000153 []
GHP1_00003018 []
GHP1_00000030 []
GHP3_00000419 ['Gymnocarpium dryopteris']
GHP1_00000874 []
GHP2_00008966 ['Chascolytrum itatiaiae', 'Briza macrostachya', 'Chascolytrum monandrum', 'Chascolytrum paleapiliferum', 'Agrostis tolucensis', 'Calamagrostis angustifolia']
GHP2_00009529 ['Juncus parryi']
GHP1_00008815 []
GHP3_00027523 []
species_name taxid
GHP2_00000044 <NA> 4209
GHP2_00000355 <NA> 102804
GHP2_00000136 <NA> 3445
GHP1_00000082 Athyrium sinense 672195
GHP2_00000549 <NA> NA
GHP1_00000014 <NA> 721813
GHP2_00000171 <NA> 981071
GHP2_00000260 <NA> 139271
GHP2_00000820 <NA> 216718
GHP1_00000361 <NA> 32109
GHP2_00000554 <NA> 4209
GHP3_00000569 <NA> 13804
GHP1_00000681 <NA> 3514
GHP3_00000153 <NA> 3514
GHP1_00003018 <NA> 3514
GHP1_00000030 <NA> 3514
GHP3_00000419 Gymnocarpium dryopteris 32116
GHP1_00000874 <NA> 1965347
GHP2_00008966 <NA> 1652080
GHP2_00009529 <NA> 13578
GHP1_00008815 <NA> 52989
GHP3_00027523 <NA> 4637
sequence
GHP2_00000044 atcacgttttccgaaaacaaacaaaggttcagaaagcgaaaataaaaaag
GHP2_00000355 atcacgttttccgaaaacaaacaaaggttcagaaagcgaaaagaaaaaaaa
GHP2_00000136 atcctgctttcagaaaacaaaaagagggttcagaaagcaaagg
GHP1_00000082 atcttgtattattcggatgaatttcgggcgatgaggcga
GHP2_00000549 atccgtgttttgagaaaacaaaggggttctcgaatcgaactataatacaaaggaaaag
GHP1_00000014 atcctgttttatgaaaataaacaagggtttcataaaccgaaaataaaaaag
GHP2_00000171 atcatgggttacgcgaacaaaccaaagtttagaaagcgg
GHP2_00000260 atccggttcatgaagacaatgtttcttctcctaagataggaaggg
GHP2_00000820 atcctgttttcccaaaacaaaggtttcaaaaaacgaaaaaaag
GHP1_00000361 atcttgtattattcagatgaatttcgggcgatgaggcga
GHP2_00000554 atcacgttttccgaaaacaaaggttcagaaagcgaaaatcaaaaag
GHP3_00000569 atcttgttttcataacataaatgg
GHP1_00000681 gtcctgttttccgaaaacaaataaaacaaatttaagggttcataaagtgagaataaaaaag
GHP3_00000153 ctcctgttttccgaaaacaaataaaacaaatttaagggttcataaagtgagaataaaaaag
GHP1_00003018 ttcctgttttccgaaaacaaataaaacaaatttaagggttcataaagtgagaataaaaaag
GHP1_00000030 tcctgttttccgaaaacaaataaaacaaatttaagggttcataaagtgagaataaaaaag
GHP3_00000419 atcttgtattactcaaatgaatttcgggcaatgaggcaa
GHP1_00000874 atcctgtttagcaaatggcgg
GHP2_00008966 atccgtgttttgagaaaacaaaggggttctcaaatcgaactataatacaaaggaaaag
GHP2_00009529 gtctttattttgataaaatttgtttttatagaaaaattcaaatcaaaaaa
GHP1_00008815 atattattttatttaaaaataa
GHP3_00027523 atccttattttgagaaaacaaaggtttataaaactagaatttaaaag
sequence_type is_spermatophyta
GHP2_00000044 SPER01 TRUE
GHP2_00000355 SPER01 TRUE
GHP2_00000136 SPER01 TRUE
GHP1_00000082 SPER01 FALSE
GHP2_00000549 SPER01 NA
GHP1_00000014 SPER01 TRUE
GHP2_00000171 SPER01 TRUE
GHP2_00000260 SPER01 TRUE
GHP2_00000820 SPER01 TRUE
GHP1_00000361 SPER01 FALSE
GHP2_00000554 SPER01 TRUE
GHP3_00000569 SPER01 FALSE
GHP1_00000681 SPER01 TRUE
GHP3_00000153 SPER01 TRUE
GHP1_00003018 SPER01 TRUE
GHP1_00000030 SPER01 TRUE
GHP3_00000419 SPER01 FALSE
GHP1_00000874 SPER01 FALSE
GHP2_00008966 SPER01 TRUE
GHP2_00009529 SPER01 TRUE
GHP1_00008815 SPER01 FALSE
GHP3_00027523 SPER01 TRUESPER01.final <- SPER01.merged4[,which(to_keep)]
SPER01.final@reads <- decostand(SPER01.final@reads,method = "total")Saving the filtered dataset
Updating the sample metadata
Adding samples metadata
metadata <- read_csv("Data/Faeces/metadata.csv",
show_col_types = FALSE)SPER01.final@samples %<>%
select(sample_id,animal_id) %>%
left_join(metadata,by = "sample_id") %>%
mutate(id = sample_id) %>%
column_to_rownames("id") %>%
select(sample_id,animal_id,Sample_number,Date,Sample_time,times_from_birch, Fed_biomass)Homogenize time from burch
Adds : - 6 hours to animal X, - 3 hours to animal Y, - 4 hours to animal 2
SPER01.final@samples %<>%
mutate(times_from_birch = times_from_birch +
ifelse(animal_id == "X",6,
ifelse(animal_id == "Y",3,4)))SPER01.final@samples %<>%
mutate(Animal_id = ifelse(animal_id == "X","9/10",
ifelse(animal_id == "Y","10/10","12/10")))Adds pellets consumption data
pellets <- read_tsv("Data/pellet_weigth.txt", show_col_types = FALSE) %>%
mutate(Date = str_replace(Date,"2018","18")) %>%
separate(Date, c("d","m","y"),sep = "/") %>%
mutate(d = as.integer(d)+1,
m = as.integer(m),
m = ifelse(d==32,m+1,m),
d = ifelse(d==32,1,d),
d = sprintf("%02d",d),
m = sprintf("%02d",m)) %>%
unite(col="Date",d,m,y,sep="/") %>%
pivot_longer(-Date,names_to = "Animal_id",values_to = "pellets")
SPER01.final@samples %<>%
left_join(pellets) Joining with `by = join_by(Date, Animal_id)`Only keep samples
SPER01.final <- SPER01.final[which(str_detect(SPER01.final@samples$sample_id,"^[XYZ]")),]Updating count statistics
SPER01.final %<>%
update_motus_count() %>%
update_samples_count() %>%
clean_empty()Add MOTUs Metadata
SPER01.final@motus %<>%
mutate(category = ifelse(is.subcladeof(taxo,taxid,spermatophyta.taxid),
"Plant",
"Lichen"))Write CSV files
write_csv(SPER01.final@samples,
file = "Data/Faeces/FE.Spermatophyta.samples.samples.csv")
write_csv(SPER01.final@motus,
file = "Data/Faeces/FE.Spermatophyta.samples.motus.csv")
write_csv(SPER01.final@reads %>%
decostand(method = "total") %>%
as.data.frame()%>%
rownames_to_column("id"),
file = "Data/Faeces/FE.Spermatophyta.samples.reads.csv")References
Wickham, H., 2016. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York.
Wickham, H., Averick, M., Bryan, J., Chang, W., McGowan, L., François, R., Grolemund, G., Hayes, A., Henry, L., Hester, J., Kuhn, M., Pedersen, T., Miller, E., Bache, S., Müller, K., Ooms, J., Robinson, D., Seidel, D., Spinu, V., Takahashi, K., Vaughan, D., Wilke, C., Woo, K., Yutani, H., 2019. Welcome to the tidyverse. Journal of open source software 4, 1686. https://doi.org/10.21105/joss.01686